News
Spatial transcriptomics — the study of all RNA molecules in a cell — is a tool that allows scientists to measure all the gene activity in a tissue and map where the activity is occurring. It is critical to understanding gene expression and the function, or malfunction, of tissues.
One of the biggest challenges in spatial transcriptomics is accurately segmenting cells to assign specific RNAs to individual cells for single-cell analysis. Today, most approaches require time-intensive manual staining or, for machine learning-enabled approaches, manually annotated datasets for model training.
Now, researchers from the Harvard John A. Paulson School of Engineering and Applied Sciences (SEAS) have developed a new computational approach which can automatically identify important biological landmarks from in situ transcriptomics. The technique incorporates the physical location and gene identity of RNAs.
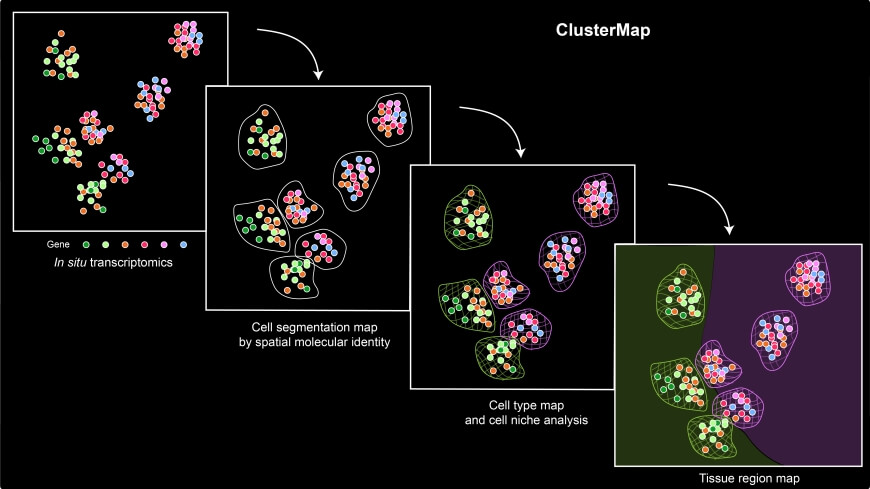
Overview of ClusterMap workflow. The input combines both spatial and transcript information of mRNA molecules sequenced by in situ transcriptomic methods. ClusterMap clusters mRNA spots, identifies cells, and profiles them into different cell types and tissue regions as output.
The framework, dubbed ClusterMap, uncovered and mapped gene expression, cell niche, and tissue region patterns from 2D and 3D spatial transcriptomics.
“This work will markedly expand our knowledge of cellular organization across all scales from subcellular organelles through cell-type maps to organs and enable further characterization of the local microenvironment for individual cells,” said Jia Liu, Assistant Professor of Bioengineering at SEAS and senior author of the paper.
The researchers demonstrate ClusterMap to be broadly applicable to various in situ transcriptomic measurements on diverse tissue types. The team envisions that, in the future, ClusterMap could be extended by combining other types of biological features, such as subcellular organelles and cell shape to uncover the basic principles of how gene expression shapes cellular architecture and tissue morphology. ClusterMap can also generate the annotated datasets for machine learning model training.
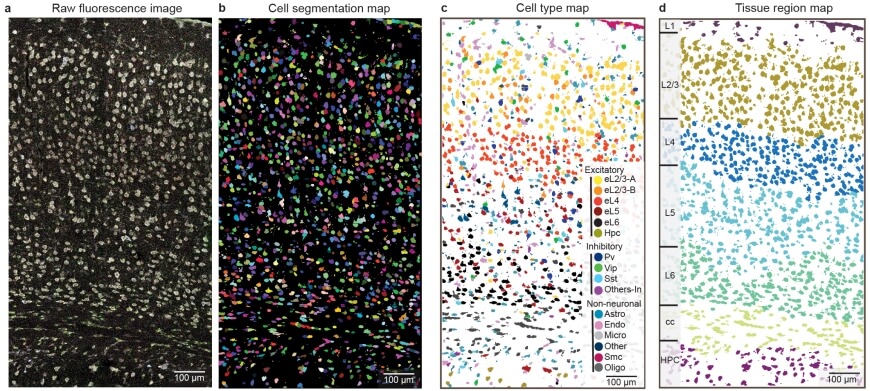
ClusterMap generated cell segmentation map (b), cell-type map (c), and tissue region map (d) of the STARmap mouse visual cortex 1020-gene dataset (a). b, mRNA molecules are color-coded by their cell attributes. c, Cells are color-coded by their cell type assignment. d, Cells are color-coded by their tissue regions.
The research was published in Nature Communications and was a collaboration with Xiao Wang’s lab at MIT and the Broad Institute. The work was led by SEAS students Yichun He and Xin Tang.
It was supported in part by the National Institutes of Health under grant 1RF1MH123948 and the National Science Foundation under grant 581ECCS-2038603.
Topics: Bioengineering
Cutting-edge science delivered direct to your inbox.
Join the Harvard SEAS mailing list.
Scientist Profiles

Jia Liu
Assistant Professor of Bioengineering
Press Contact
Leah Burrows | 617-496-1351 | lburrows@seas.harvard.edu


